Chemodectoma (paraganglioma): eziologia, patogenesi, classificazione, diagnostica, metodi di trattamento.
Kizyukevich O.Chirurgo cardiovascolare, MD
10 minuti di lettura·Maggio 07, 2025
Questo articolo è solo a scopo informativo
Il contenuto di questo sito web, inclusi testi, grafici e altri materiali, è fornito solo a scopo informativo. Non sono da intendersi come consigli o indicazioni. Per quanto riguarda la tua specifica condizione medica o il tuo trattamento, ti invitiamo a consultare il tuo operatore sanitario.
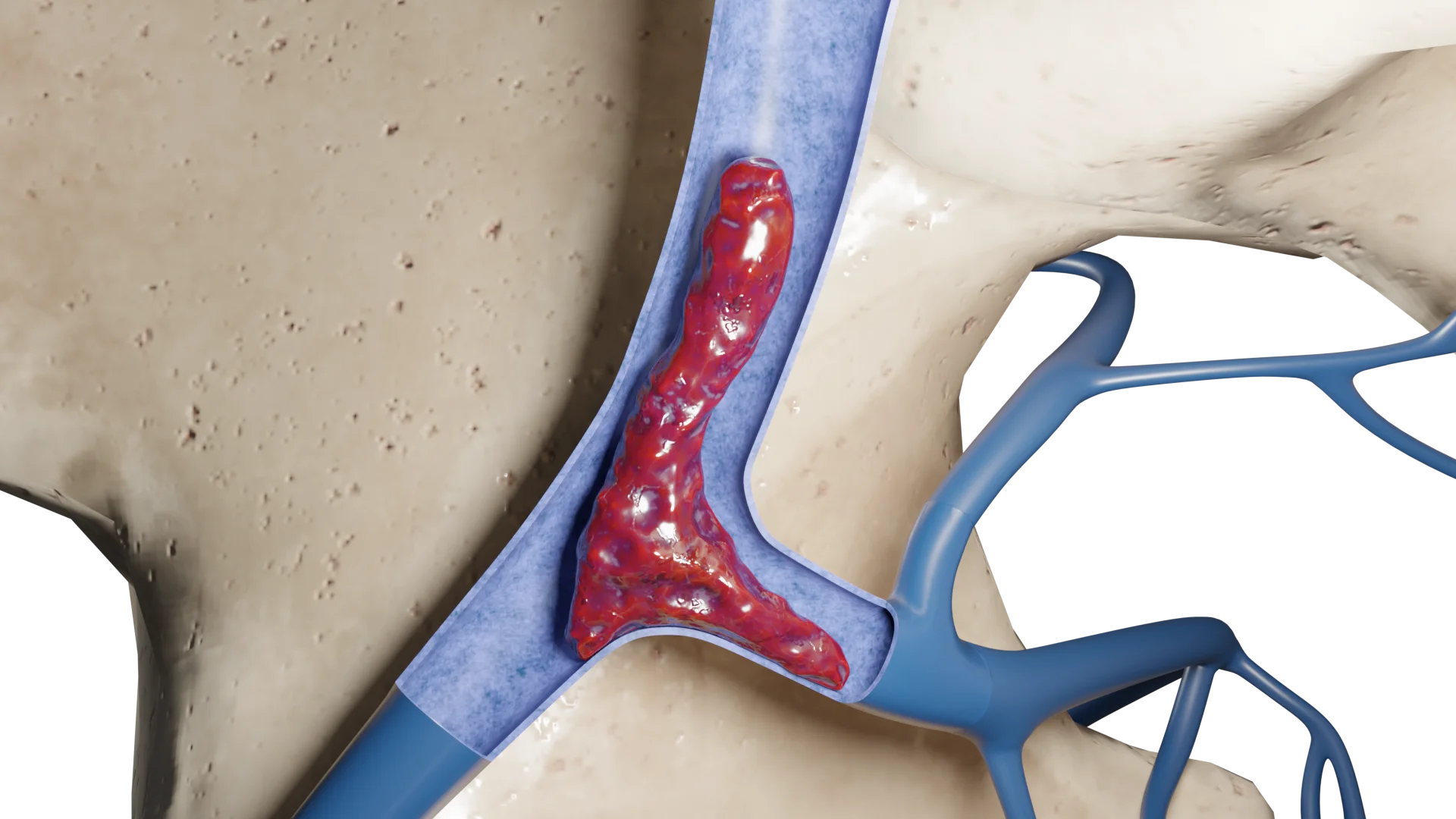
Il paraganglioma del corpo carotideo (chemodectoma) è un tumore raro, prevalentemente benigno, originato dalle cellule chemocettive del corpo carotideo, situato nella biforcazione della carotide comune.
Le donne sono più spesso colpite dalla malattia rispetto agli uomini.
L’età media alla diagnosi è circa 45 anni.
L’elevata prevalenza è osservata nei paesi dell’America Latina, specialmente in Messico, dove le donne rappresentano fino al 90% dei casi.
Gli abitanti delle regioni di alta montagna hanno un aumento del rischio di sviluppare chemodectomi a causa dell’ipossia cronica.
Eziologia
Mutazioni genetiche — giocano un ruolo predominante le mutazioni nei geni che codificano per le subunità del complesso succinato deidrogenasi (SDH), che coinvolge la catena respiratoria dei mitocondri e il metabolismo cellulare. Principali geni:
SDHD — più comunemente associato a paragangliomi multipli di testa e collo; è trasmesso per linea paterna;
SDHB — associato a forme più aggressive e con rischio aumentato di comportamento maligno;
SDHC, SDHA, SDHAF2 — meno frequentemente coinvolti ma possono anche contribuire allo sviluppo del tumore.
Sindrome, associata con chemodectomi:
sindrome di Von Hippel-Lindau (VHL);
neoplasia endocrina multipla di tipo 2 (MEN2);
neurofibromatosi di tipo 1 (NF1).
Ipossia cronica — vivere a un’altitudine superiore a 2000 metri sul livello del mare, insieme a condizioni come la broncopneumopatia cronica ostruttiva e difetti congeniti del cuore, può stimolare l’iperplasia del corpo carotideo.
Storia familiare — circa il 10 % dei casi è familiare.
Animazione 3D – chemodectoma I tipo
Animazione 3D – chemodectoma III tipo
Patogenesi
Mutazioni ereditarie (SDHx)
Mutazioni SDH (enzima chiave del complesso II della catena respiratoria mitocondriale e ciclo di Krebs) → accumulo di succinato (funziona come oncometabolite) → inattivazione delle proliidrossilasi (PHDs) — enzimi che controllano la degradazione di HIF-1α (fattore inducibile da ipossia) → accumulo di HIF-1α → pseudohipossia → attivazione di VEGF (angiogenesi) / GLUT1 (glicolisi) / PDGF (proliferazione cellulare) → trasformazione neoplastica delle cellule chemocettive e formazione del tumore.
Ipossia cronica
Ipossia (altitudine, BPCO) → riduzione di pO₂ → attivazione dei recettori carotidi → iperplasia delle cellule → accumulo di HIF-1α → angiogenesi e neoplasia.
Di conseguenza, il tumore cresce progressivamente di dimensioni. Durante la crescita può mostrare un carattere invasivo locale, fino a circondare o comprimere le strutture anatomiche vicine come arterie carotidi interna ed esterna, nervi vaghi, ipoglossi e glossofaringei. Questo può causare corrispondente sintomatologia neurologica e vascolare.
Classificazione del paraganglioma
William Shamblin propose di dividere i paragangliomi del corpo carotideo in tre tipi (classificazione Shamblin):
tipo I: tumori limitati fino a 3,5 cm, debolmente associati alle pareti delle arterie;
tipo II: 3,5-5 cm, coprono parzialmente le arterie carotidi, hanno un’aderenza più densa alle pareti delle arterie;
tipo III: più di 5 cm, coprono le arterie carotidi e/o i vasi e nervi vicini con un’adherenza notevolmente densa a questi strutture.
Inoltre, il chemodectoma può essere classificato per eziologia: sporadica (fino all’85 %), familiare (10–15 %), iperplastica (1–5 %).
Chemodectoma tipo I
Chemodectoma tipo III
Manifestazioni Cliniche
Per lungo tempo i sintomi possono essere assenti. Man mano che il tumore cresce, possono comparire lamentele:
tumore palpabile — massa lentamente crescente, indolore e pulsante sulla superficie laterale del collo (nella regione del muscolo sternocleidomastoideo);
sintomi neurologici — raucedine, disfagia, intorpidimento della lingua, causati dalla compressione dei nervi cranici (IX–XII);
disturbi legati alla produzione di catecolamine — palpitazioni, episodi di ipertensione arteriosa, sudorazione, mal di testa, tremore;
manifestazioni rare — vertigini, svenimenti per compressione del seno carotideo.
Diagnostica del paraganglioma
Esame obiettivo: tumore denso e pulsante sul collo, che si sposta orizzontalmente ma non verticalmente.
Ecografia con doppler: tumore ipervascolare nella biforcazione della carotide.
RM con contrasto: aspetto caratteristico “sale e pepe” nelle immagini pesate in T1.
TC con contrasto: valutazione del grado di invasione nei vasi e classificazione secondo Shamblin. Costruzione di un modello 3D per la pianificazione chirurgica.
Angiografia: rilevazione del “segno della lira” — divergenza delle arterie carotide interne ed esterne. Permette di valutare la necessità/possibilità di embolizzazione dell’area interessata come fase preoperatoria.
PET-TC con 68Ga-DOTATATE: in caso di sospetto di focolai multipli o metastatici.
Esami di laboratorio: determinazione dei livelli di metanefrine e normetanefrine nel plasma e nelle urine se si sospetta un tumore secernente.
Test genetici per mutazioni nei geni SDH in presenza di anamnesi familiare o tumori multipli.
Scopri altri contenuti scientificamente accurati sui nostri social network
Iscriviti e non perdere le ultime risorse
Trattamento
Il trattamento della chemodectomia carotidea dipende dalle dimensioni del tumore, dalla sintomatologia clinica, dal rischio di complicazioni, dall’attività funzionale, dal profilo genetico e dalla classe Shamblin. I principali metodi di trattamento sono la rimozione chirurgica, l’embolizzazione preoperatoria e, in alcuni casi, la radioterapia o la chemioterapia.
Embolizzazione preoperatoria
L’embolizzazione preoperatoria viene eseguita per ridurre la vascolarizzazione del tumore, diminuire il volume della perdita ematica intraoperatoria e semplificare l’asportazione del tumore durante la chirurgia.
Indicazioni:
tumori di classe Shamblin II–III;
diametro del tumore > 3 cm;
presenza di un flusso arterioso significativo (secondo i dati TC/angiografici);
resezione pianificata con ricostruzione vascolare.
Controindicazioni:
tumori di classe Shamblin I;
assenza di componente arterioso (bassa vascolarizzazione);
anastomosi con la circolazione cerebrale — rischio di embolizzazione del cervello.
Si esegue un’angiografia selettiva dell’arteria carotide esterna. Attraverso un microcatetere si iniettano agenti embolizzanti: particelle di alcool polivinilico (PVA), microsfere, meno frequentemente colla o spirali. L’angiografia di controllo conferma la riduzione del flusso sanguigno. L’intervento viene eseguito entro 24–48 ore dall’embolizzazione, finché l’effetto persiste.
mutazione confermata di SDHB (rischio di malignità);
età < 60 anni.
Tipi di trattamento chirurgico:
Resezione extracapsulare del tumore. Standard per classe Shamblin I–II, conservazione dei vasi, rischio minimo.
Resezione della carotide con ricostruzione. Più comune con classe Shamblin III. Dopo la resezione della sezione dell’arteria carotide, viene eseguita una protesizzazione (Dacron, PTFE).
Neuromonitoraggio e microchirurgia. Utilizzato quando vicino ai nervi craniali (IX–XII).
Possibili complicanze:
emorragia (soprattutto se l’embolizzazione è insufficiente);
danni ai nervi craniali (IX–XII) — fino al 30% nei tumori di grandi dimensioni;
ictus (se si compromette il flusso sanguigno collaterale);
recidiva (raro, in caso di resezione incompleta).
Chemioterapia
Non è il metodo standard di trattamento per la chemodectomia carotidea, poiché la maggior parte dei tumori cresce lentamente e ha una natura benigna. Tuttavia, nei rari casi di decorso maligno (progressione rapida, coinvolgimento metastatico) o in forme metastatiche non operabili può essere utilizzata una terapia sistemica.
Terapia radionucleare ¹⁷⁷Lu-DOTATATE (PRRT)
Assegnata in presenza di espressione dei recettori della somatostatina tramite PET con ⁶⁸Ga-DOTATATE.
FAQ
1. La chemodectomia carotidea è un tumore maligno?
Nella maggior parte dei casi — no. La chemodectomia carotidea appartiene ai tumori benigni a crescita lenta. Tuttavia, in presenza di mutazioni del gene SDHB, può esserci un potenziale maligno con metastasi.
2. Si può semplicemente osservare una chemodectomia carotidea senza rimuoverla?
Sì, in alcuni casi è possibile un’osservazione attiva, soprattutto se il tumore è di piccole dimensioni (< 2,5 cm), asintomatico, in pazienti anziani o se ci sono controindicazioni all’intervento chirurgico. Tuttavia, in presenza di segni di crescita o compressione delle strutture del collo, è necessario il trattamento.
3. L’operazione per la rimozione della chemodectomia carotidea è pericolosa?
I rischi dipendono dalle dimensioni e dalla posizione del tumore. In grandi formazioni possono verificarsi complicazioni, inclusi danni ai nervi craniali, emorragie ed eventi ischemici. Per ridurre i rischi, spesso viene eseguita un’embolizzazione preoperatoria.
4. È necessario esaminare altri membri della famiglia se mi è stata diagnosticata una chemodectomia?
Se viene rilevata una mutazione ereditaria (ad esempio, SDHD, SDHB), si consiglia la consulenza genetica e l’esame dei parenti stretti, poiché la malattia può essere familiare.
5. Come distinguere la chemodectomia carotidea da altri tumori del collo?
La chemodectomia carotidea si localizza di solito nella zona di biforcazione dell’arteria carotide comune, pulsa, si sposta orizzontalmente ma non verticalmente e mostra il caratteristico “segno della lira” alla TC/RMN. La diagnosi viene confermata tramite imaging e angiografia.
6. Una chemodectomia può causare dolore o disagio?
Nella maggior parte dei casi — no, specialmente negli stadi iniziali. Tuttavia, con la crescita, il tumore può esercitare pressione su nervi e vasi, causando dolore, raucedine, disturbi della deglutizione o vertigini.
7. Esiste il rischio di recidiva dopo la rimozione del tumore?
In caso di rimozione completa del tumore, il rischio di recidiva è basso (meno del 5%). Tuttavia, in caso di resezione incompleta, forma ereditaria o presenza di mutazione SDHB, è possibile una recidiva o lo sviluppo di nuovi focolai. In tali casi è importante l’osservazione a lungo termine.
Bibliografia
1.
VOKA 3D Anatomy & Pathology – Complete Anatomy and Pathology 3D Atlas (VOKA Anatomia e Patologia 3D – Atlante 3D completo di anatomia e patologia) [Internet]. VOKA 3D Anatomy & Pathology (VOKA Anatomia e patologia 3D).
Disponibile su: https://catalog.voka.io/
2.
Luna-Ortiz K, Reynoso-Noverón N, Herrera-Ponzanelli C, Favila-Lira S, Luna-Peteuil Z, Herrera-Gomez A, et al. Differenze sessuali nella presentazione etnica dei tumori del corpo carotideo: una revisione sistematica della letteratura. Int J Otorhinolaryngol Head Neck Surg. 2022 Jun;8(6):527-531. doi: 10.18203/issn.2454-5929.ijohns20221393.
3.
Butt N, Baek WK, Lachkar S, Iwanaga J, Mian A, Blaak C, et al. Il corpo carotideo e i tumori associati: revisione aggiornata con significato clinico/chirurgico. Br J Neurosurg. 2019 Oct;33(5):500-503. doi: 10.1080/02688697.2019.1617404.
4.
Darouassi Y, Alaoui M, Mliha Touati M, Al Maghraoui O, En-Nouali A, Bouaity B, Ammar H. Tumori del corpo carotideo: una serie di casi e revisione della letteratura. Ann Vasc Surg. 2017 Ago;43:265-271. doi: 10.1016/j.avsg.2017.03.167.
5.
Gonzalez-Urquijo M, Castro-Varela A, Barrios-Ruiz A, Hinojosa-Gonzalez DE, Salas AKG, Morales EA, et al. Tendenze attuali nei tumori del corpo carotideo: revisione completa. Head Neck. 2022 Oct;44(10):2316-2332. doi: 10.1002/hed.27147.
6.
Ozawa H. Gestione attuale dei tumori del corpo carotideo. Auris Nasus Larynx. 2024 Jun;51(3):501-506. doi: 10.1016/j.anl.2024.01.007.
7.
Piazza C, Lancini D, Tomasoni M, Zafereo M, Poorten VV, Hanna E, et al. Tumori maligni del corpo carotideo: cosa sappiamo, cosa facciamo e cosa dobbiamo ottenere. Una revisione sistematica della letteratura. Head Neck. 2024 Mar;46(3):672-687. doi: 10.1002/hed.27624.