Ependimomas: etiología, clasificación, diagnóstico, tratamiento y pronóstico
Dobriyan S.Oncólogo quirúrgico, MD
17 min leer·noviembre 13, 2025
Este artículo sólo tiene fines informativos
El contenido de este sitio web, incluidos textos, gráficos y otros materiales, se proporciona únicamente con fines informativos. No pretende ser un consejo u orientación. En relación con tu enfermedad o tratamiento específico, consulta a tu médico.
Los ependimomas son un grupo de tumores gliales que se originan en las células del tejido ependimario que recubre el sistema ventricular del cerebro y el canal espinal. Estos tumores pueden localizarse en diversas partes del sistema nervioso central, con mayor frecuencia en la fosa craneal posterior y la médula espinal. El enfoque moderno para el tratamiento de los ependimomas se basa en la extirpación quirúrgica máxima del tumor seguida de radioterapia. Las características anatómicas, histológicas y genéticas moleculares del tumor influyen en el pronóstico.
Las causas exactas de la aparición de los ependimomas no se han establecido.
Existen varios mecanismos que provocan el desarrollo de este tumor:
Alteraciones genéticas: los ependimomas a menudo presentan alteraciones en la estructura del ADN —desequilibrios cromosómicos (p. ej., alteración cromosómica +1q), fusiones de genes (p. ej., fusiones ZFTA-RELA), deleciones y mutaciones— que conducen a la activación de vías oncogénicas y a la supresión de los mecanismos de control del crecimiento celular.
Cambios epigenéticos (cambios en la actividad genética): la desregulación de la metilación del ADN y de las histonas es particularmente importante, por ejemplo, la pérdida de la trimetilación de H3K27me3 en el grupo PF-EPN-A, que suprime la expresión de genes responsables de la diferenciación.
Influencia de factores hereditarios: existe una asociación entre algunos ependimomas medulares y la schwannomatosis asociada a NF2.
Por lo tanto, el ependimoma resulta de una interacción compleja de procesos genéticos y epigenéticos que conducen a la alteración del control celular y al crecimiento proliferativo excesivo.
Epidemiología
Los ependimomas se diagnostican a cualquier edad, aunque la incidencia máxima se produce en la primera infancia (mediana de 5 años).
En la infancia, el 90% de los ependimomas se localizan en la cabeza, principalmente en la fosa craneal posterior.
En los adultos, la incidencia general de ependimomas es menor; se observan más casos en la médula espinal (65%).
Manifestaciones clínicas
Los síntomas del ependimoma dependen de su ubicación.
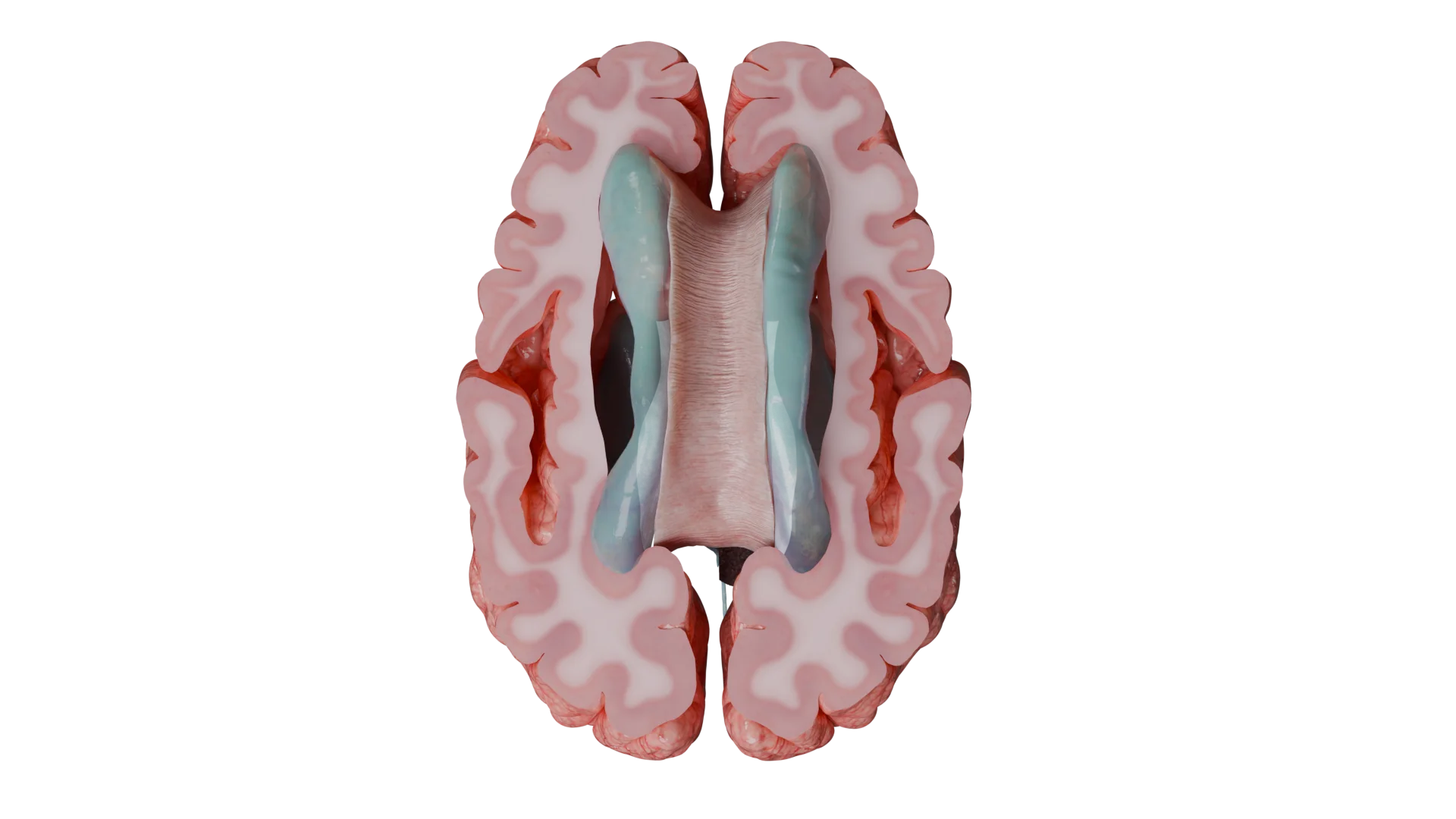
Tumores supratentoriales (por encima de la tienda del cerebelo): se caracterizan por convulsiones, manifestaciones neurológicas focales (trastornos del habla, cambios de personalidad y comportamiento, disminución de la memoria y la atención, hemiparesia) y síntomas cerebrales generales (dolor de cabeza, náuseas, vómitos, papiledema).
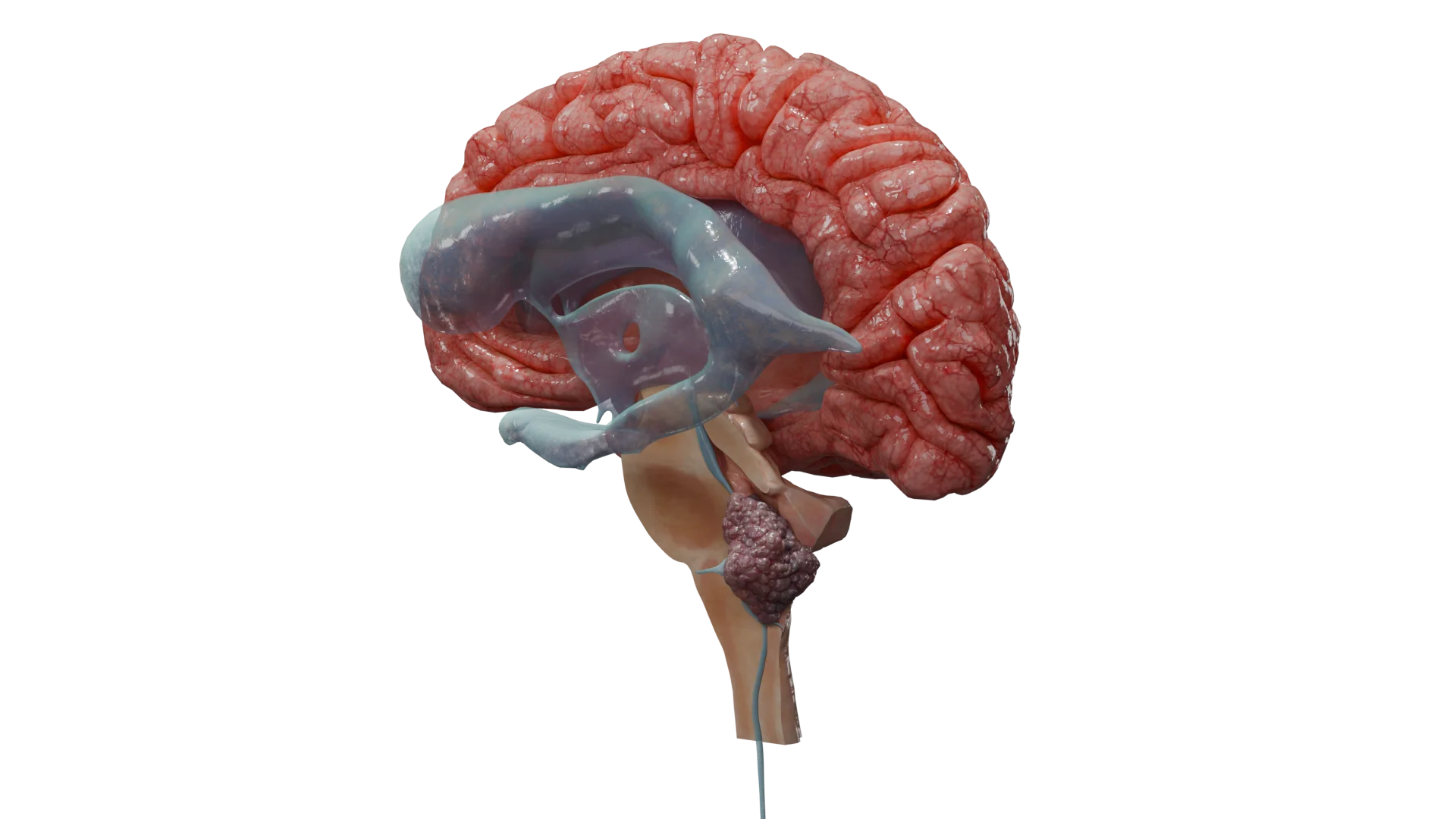
Ventrículos laterales: un sitio típico para la aparición de ependimomas supratentoriales
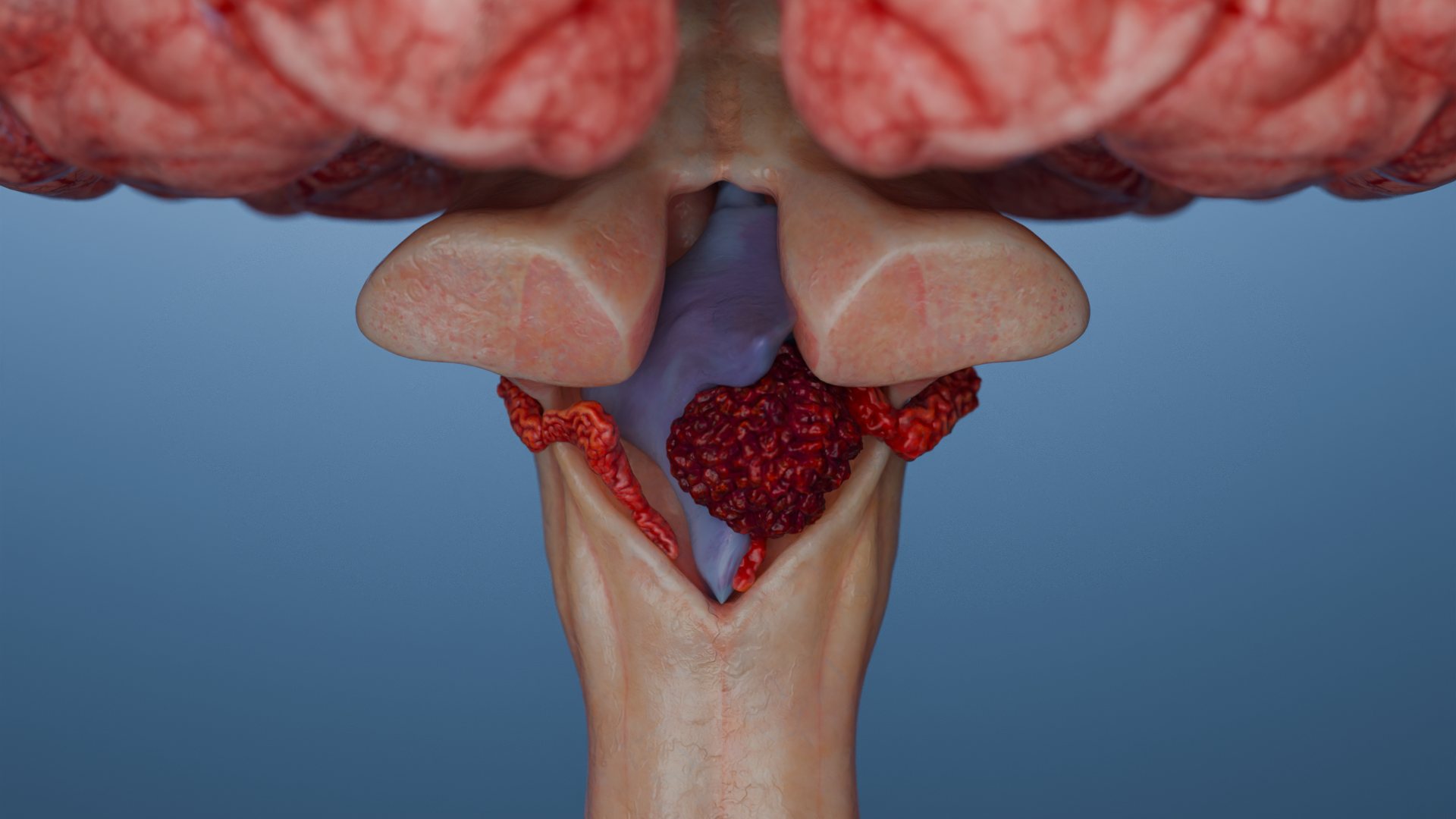
Tumores infratentoriales (en la fosa craneal posterior): se acompañan de los signos de aumento de la presión intracraneal (hidrocefalia – dolor de cabeza, náuseas, vómitos), la ataxia (deterioro de la coordinación de los movimientos e inestabilidad) o la disfunción de los nervios craneales (disartria, disfagia, pérdida auditiva).
Animación 3D: Ependimoma infratentorial
Ependimomas medulares: se manifiestan con dolor de espalda, dolor radicular, debilidad muscular creciente, disfunción pélvica (trastornos de la micción y la defecación, a veces impotencia).
Lugar de aparición de los ependimomas medulares
Clasificación de los ependimomas
Los ependimomas se clasifican en función de sus características anatómicas, histológicas y moleculares.
En 2021, la OMS introdujo cambios importantes en la clasificación de los ependimomas, destacando nuevas características moleculares de importancia para el pronóstico y el tratamiento personalizado.
Los datos moleculares complementan significativamente la clasificación histológica tradicional de los ependimomas y permiten un tratamiento personalizado y un mejor pronóstico.
Clasificación anatómica con características moleculares (OMS 2021)
Según la clasificación de tumores del SNC de la OMS de 2021, existen 10 tipos de tumores ependimarios:
Ependimoma supratentorial (ST-EPN):
Ependimoma supratentorial, no especificado (ST-EPN-SAI/NCOP);
Ependimoma supratentorial, positivo para una fusión de ZFTA;
Ependimoma supratentorial, positivo para una fusión de YAP1.
Ependimoma de fosa posterior (PF-EPN):
Ependimoma de fosa posterior, no especificado (PF-EPN-SAI/NCOP);
Ependimoma de fosa posterior del grupo A (PFA);
Ependimoma de fosa posterior del grupo B (PFB).
Ependimoma medular (SP-EPN):
Ependimoma medular;
Ependimoma medular con amplificación de MYCN.
Ependimoma mixopapilar (MEPN). Se localiza con mayor frecuencia en la parte caudal de la médula espinal.
Subependimoma (SubEPN). Se encuentra con mayor frecuencia en el cuarto ventrículo y en los ventrículos laterales.
Clasificación de los ependimomas y sus características
Tipo de tumor
Grado según la OMS
Características moleculares clave
Características clínicas y pronóstico
Ependimoma supratentorial, positivo para una fusión de ZFTA (ST-EPN-ZFTA*)
*anteriormente: positivo para una fusión de RELA
–
Fusión de los genes ZFTA con RELA y otros genes, lo que conduce a la activación de la vía NF-κB y al crecimiento tumoral
Común en niños, el subtipo agresivo, se asocia con un mal pronóstico
Ependimoma supratentorial, positivo para una fusión de YAP1 (ST-EPN-YAP1)
–
Fusión de YAP1
Menos frecuente, común en lactantes, pronóstico más favorable
Ependimoma supratentorial, no especificado (ST-EPN-SAI/NCOP)
2 o 3
Mutaciones heterogéneas; tumor no clasificado (NCOP) o sin especificación molecular (SAI)
Grupo heterogéneo; el pronóstico y la terapia requieren más investigación
Ependimoma de fosa posterior del grupo A (PF-EPN-A)
–
Pérdida de H3K27me3; frecuentemente 1q+
Se presenta predominantemente en lactantes y niños, el subtipo agresivo con mal pronóstico
Ependimoma de fosa posterior del grupo B (PF-EPN-B)
–
Inestabilidad cromosómica, retención de H3K27me3
En los pacientes de mayor edad (adolescentes y adultos), buen pronóstico
Ependimoma de fosa posterior, no especificado (PF-EPN-SAI/NCOP)
2 o 3
Morfológicamente, el ependimoma de fosa posterior, no clasificado (NCOP) o sin especificación molecular (SAI)
El diagnóstico se basa en la localización y las características histológicas clásicas; el pronóstico es individual y depende de la extensión de la resección y la evolución clínica
Ependimoma medular
2 o 3
No existen marcadores moleculares claros, a menudo se observan pérdidas cromosómicas en 22q (donde se encuentra el gen NF2)
Pronóstico intermedio, cirugía + radioterapia
Ependimoma medular con amplificación de MYCN (SP-EPN-MYCN)
–
Amplificación de MYCN
Subtipo muy agresivo con mal pronóstico
Ependimoma mixopapilar (MEPN)
2
Diferencias entre adultos y niños
Se encuentra con mayor frecuencia en la parte caudal de la médula espinal; se observan las recaídas locales; la recuperación es posible después de la cirugía y la radioterapia
Subependimoma (SubEPN)
1
Generalmente sin mutaciones agresivas, es benigno, posible mutación en TERT
Cualquier parte del sistema ventricular y la médula espinal, crecimiento lento, a menudo un hallazgo inesperado, pronóstico favorable
Principio de la OMS para determinar el grado de malignidad (WHO Grade)
Según el grado de malignidad, los ependimomas suelen tener un grado II–III según la OMS. Cuanto mayor sea el grado, mayor será la malignidad del tumor y peor el pronóstico para el paciente.
Según la actualización de la clasificación de la OMS de 2021 para tumores del SNC, no se asigna ningún grado a los nuevos ependimomas definidos molecularmente, como los positivos para una fusión de YAP1 (ST-EPN-YAP1), los positivos para una fusión de ZFTA (ST-EPN-ZFTA), PF-A y PF-B. Se recomienda que el grado (grado de malignidad 1, 2 o 3) se indique únicamente para los ependimomas clásicos (incluidos los definidos morfológicamente, pero no especificados molecularmente: «NCOP/SAI»).
Definiciones de SAI y NCOP:
SAI (sin otra indicación): este término se utiliza cuando no se han realizado (o no se pueden realizar) todas las pruebas moleculares/genéticas u otras pruebas adicionales necesarias para el tumor, por lo que el diagnóstico se establece únicamente sobre la base de criterios básicos (morfológicos). Tomando como ejemplo el ependimoma: si un tumor cerebral coincide con la histología clásica del ependimoma, pero no se determinó el subgrupo molecular (p. ej., fusión de ZFTA, fusión de YAP1, PF-A, PF-B) o el análisis no fue adecuado para realizar un diagnóstico definitivo, se indica «ependimoma supratentorial, SAI» o «ependimoma de fosa posterior, SAI».
NCOP (no clasificado en otra parte): este término se utiliza cuando un tumor ha sido completamente caracterizado y se han realizado todas las pruebas actuales, pero no encaja en ninguno de los subtipos o grupos estandarizados conocidos. En el caso del ependimoma, si el tumor cumple los criterios para ependimoma, se ha realizado un estudio morfológico y molecular exhaustivo, pero el tumor no corresponde a ninguno de los subtipos moleculares descritos (por ejemplo, no se ajusta a ZFTA, YAP1 o PF-A/PF-B), el diagnóstico será «ependimoma, NCOP».
El ependimoma hace metástasis dentro del sistema nervioso central, propagando células alteradas a través de la circulación del líquido cefalorraquídeo.
Diagnóstico de los ependimomas
Imágenes por resonancia magnética (IRM)
La base del diagnóstico es la resonancia magnética del cerebro y la médula espinal con contraste.
Los ependimomas supra e infratentoriales suelen contener calcificaciones y componentes quísticos y se caracterizan por hemorragia y realce heterogéneo en la RM.
En las imágenes, los ependimomas suelen ser hipointensos en T1 e hiperintensos en T2, a menudo con un marcado realce de contraste.
Los ependimomas medulares se calcifican con menor frecuencia y pueden presentar una señal hipointensa en T2 debido al depósito de hemosiderina, lo que se conoce como el “signo de la copa”.
Los ependimomas mixopapilares suelen ser isointensos en T1 e hiperintensos en T2 en comparación con los tumores de la médula espinal.
Citología del líquido cefalorraquídeo
La citología del líquido cefalorraquídeo es importante para el estadiaje y se realiza con mayor frecuencia después de la cirugía, incluso para determinar el alcance de la radioterapia postoperatoria.
Examen histológico
La confirmación histológica es obligatoria al extirpar este tumor. Se examina bajo un microscopio una muestra del tumor obtenida durante la cirugía para determinar su tipo y grado de malignidad.
Diagnóstico diferencial
El diagnóstico depende de la ubicación:
En la región supratentorial: gliomas, tumores embrionarios y tumores del plexo coroideo.
En la fosa craneal posterior: meduloblastoma, astrocitoma pilocítico y tumores del plexo coroideo.
En la médula espinal: astrocitoma medular, schwannoma y meningioma.
Tratamiento de los ependimomas
Tratamiento quirúrgico
La extirpación quirúrgica es el tratamiento principal para los ependimomas.
La extirpación completa del tumor es un factor crítico para el pronóstico. La cirugía requiere alta calificación del médico debido a la proximidad a estructuras vitales.
En ocasiones, se realizan operaciones de derivación para la hidrocefalia con el fin de prevenir la acumulación excesiva de líquido cefalorraquídeo en los ventrículos cerebrales.
Debido a la ubicación del ependimoma, la extirpación quirúrgica radical suele ser difícil de lograr, ya que se asocia con un alto riesgo de complicaciones. En estos casos, aumenta la necesidad de radioterapia postoperatoria.
Radioterapia
La radioterapia postoperatoria mejora la supervivencia libre de progresión.
Generalmente está indicada en casos de la extirpación subtotal del tumor.
En caso de diseminación, se recomienda la radioterapia craneoespinal.
Los métodos especiales de radioterapia, como radioterapia de intensidad modulada (IMRT) y terapia de protones, minimizan el daño al tejido sano.
Quimioterapia
La quimioterapia tiene una eficacia limitada y se utiliza principalmente en niños menores de 1–1,5 años para retrasar la radioterapia o en casos de recaída y cuando no es posible realizar cirugía o radiación. Los medicamentos quimioterapéuticos utilizados incluyen cisplatino, carboplatino, ciclofosfamida, etopósido y metotrexato.
En niños mayores de 1 año y en adultos, el impacto de la quimioterapia es limitado debido a la baja sensibilidad de los ependimomas a la mayoría de los citostáticos. En raras ocasiones, puede recomendarse en casos de recaída o cuando el tratamiento quirúrgico y la radioterapia no son posibles.
Terapia dirigida e inmunoterapia
Existen datos experimentales sobre la terapia dirigida basada en perfiles moleculares (por ejemplo, inhibidores de EGFR y VEGF), pero su aplicación clínica es limitada y requiere más investigación.
Encuentra más contenido científicamente preciso en nuestras redes sociales
Suscríbete y no te pierdas los últimos recursos
Pronóstico y observación
La supervivencia depende de la edad del paciente, la extensión de la resección, el subtipo molecular del tumor y la presencia de diseminación (propagación).
Con resección completa y terapia adecuada, la tasa de supervivencia a 5 años en niños es superior al 70%.
En pacientes adultos, el pronóstico también depende del tipo de tumor y generalmente es favorable con la extirpación total y la posterior radioterapia.
Es necesario un seguimiento a largo plazo con resonancia magnética regular durante al menos 5 años para detectar recaídas tardías.
FAQ
1. ¿Qué es un ependimoma y dónde se puede localizar?
El ependimoma es un tumor glial del sistema nervioso central que se origina en las células que recubren los ventrículos cerebrales y el canal central de la médula espinal. En los niños, hasta el 90% de los ependimomas se localizan en el cerebro (principalmente en la fosa craneal posterior), mientras que en los adultos, hasta el 65% de los casos se producen en la médula espinal.
2. ¿Cuáles son los síntomas del ependimoma?
Los síntomas del ependimoma dependen directamente de la ubicación del tumor. Cuando el ependimoma afecta al cerebro, pueden aparecer signos de aumento de la presión intracraneal (dolor de cabeza, náuseas), convulsiones, ataxia (deterioro de la coordinación) y déficit neurológico focal. El ependimoma de la médula espinal, incluyendo la zona de la cauda equina, se caracteriza por dolor de espalda, debilidad muscular en las piernas y disfunción pélvica (problemas con la micción y la defecación).
3. ¿El ependimoma es maligno o benigno?
Los ependimomas son un grupo amplio de tumores. Existen tipos benignos de crecimiento lento (por ejemplo, el subependimoma de grado I según la OMS), pero la mayoría de los ependimomas son de grado II o III y se consideran malignos. La clasificación actual de la OMS hace hincapié en las características moleculares que determinan con mayor precisión el grado de malignidad y el pronóstico de la enfermedad.
4. ¿Cómo se trata el ependimoma?
El tratamiento se basa en la extirpación quirúrgica completa del tumor. Dado que la extirpación radical del tumor no siempre es posible, el segundo paso clave es la radioterapia postoperatoria, que mejora significativamente el control de la enfermedad. El impacto de la quimioterapia es limitado: se utiliza principalmente en niños pequeños para retrasar la radiación, y también en casos de recaídas cuando se han agotado otros métodos.
5. ¿Cuál es el pronóstico del ependimoma y cuánto tiempo viven los pacientes?
El pronóstico y la supervivencia varían ampliamente y dependen del subtipo molecular del tumor, la edad del paciente y, lo que es más importante, la integridad de la extirpación quirúrgica. Con la resección total y la posterior radioterapia, la tasa de supervivencia a 5 años en niños supera el 70%. El pronóstico en adultos también suele ser favorable, siempre que se realice el tratamiento adecuado. Sin embargo, los subtipos moleculares agresivos, como el PF-EPN-A en niños o el ependimoma medular con amplificación de MYCN, tienen un peor pronóstico.
Bibliografía
1.
Catálogo VOKA. [Recurso electrónico]
https://catalog.voka.io/
2.
Upadhyaya SA, Tinkle C. Intracranial ependymoma and other ependymal tumors [Ependimoma intracraneal y otros tumores ependimarios]. UpToDate. 2023
3.
Kresbach C, Neyazi S, Schüller U. (2022). Updates in the classification of ependymal neoplasms: The 2021 WHO Classification and beyond [Actualizaciones en la clasificación de las neoplasias ependimarias: La clasificación de la OMS de 2021 y más allá]. Brain Pathol. Julio de 2022;32(4):e13068. doi: 10.1111/bpa.13068. Epub 21 de marzo de 2022. PMID: 35307892; PMCID: PMC9245931.
4.
Mu W, Dahmoush H. (2023). Classification and neuroimaging of ependymal tumors [Clasificación y neuroimagen de los tumores ependimarios]. Front Pediatr. 23 de mayo de 2023;11:1181211. doi: 10.3389/fped.2023.1181211. PMID: 37287627; PMCID: PMC10242666.
5.
Mack SC, Witt H, Pajtler KW et al. (2018). Therapeutic targeting of ependymoma as informed by oncogenic enhancer profiling [Tratamiento dirigido del ependimoma según lo informado por el perfil de potenciadores oncogénicos]. Nature. 2018;553(7686):101-5.
6.
Parker M et al. (2014). C11orf95-RELA fusions drive oncogenic NF-kB signalling in ependymoma [Las fusiones C11orf95-RELA impulsan la señalización oncogénica de NF-κB en el ependimoma]. Nature. 2014;506(7489):451-5.
7.
Yamaguchi J et al. (2023). Latest classification of ependymoma in the molecular era and advances in its treatment: a review [Clasificación más reciente de los ependimomas en la era molecular y avances en su tratamiento: una revisión]. Jpn J Clin Oncol. 2023;53(8):653-663.
8.
Merchant TE et al. (2009). Conformal radiotherapy after surgery for paediatric ependymoma: overall survival and toxicity [Radioterapia conformada tras cirugía de ependimoma pediátrico: supervivencia global y toxicidad]. Lancet Oncol. 2009;10(3):258-66.
9.
National Comprehensive Cancer Network (NCCN) CNS Cancers Guidelines [Guías de la Red Nacional Integral del Cáncer (NCCN) sobre cánceres del SNC]. (2024).
10.
Tsang DS et al. (2018). Reirradiation for recurrent pediatric intracranial ependymoma [Reirradiación para el ependimoma intracraneal pediátrico recurrente]. Int J Radiat Oncol Biol Phys. 2018;100(2):507-514.
Petersburg FL 33702, 7901 4th St N STE 300, ESTADOS UNIDOS
¡Gracias!
¡Tu mensaje ha sido enviado! Nuestros expertos se pondrán en contacto contigo en breve. Si tienes más preguntas, ponte en contacto con nosotros en info@voka.io.